
Sindrome de Goltz [Dataset]
Mostra el registre complet de l'element
|
|
|
|
Vera Sempere, Francisco José
|
|
Aquest document és un/a DataSet, creat/da en: 2017
|
|
|
Tipo: dataset
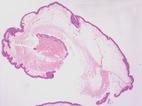
Resumen: El síndrome reconocido con la denominación
eponemica de síndrome de Goltz o síndrome de Goltz-Gorlin (no
confundir con el síndrome de Gorlin-Goltz), es también denominado
Hipoplasia dérmica focal. Este síndrome fue descrito clínicamente en
1962 siendo identificado el gen implicado en su génesis en el año
2007.Se trata de una rara afección de trasmisión genética, ligada al
cromosoma X, siendo referida en el catálogo de enfermedades de
trasmisión genética con el código OMIM# 305600. Se constituye como una
enfermedad rara (ORPHA 2092) y aunque se desconoce su exacta
prevalencia, esta previsiblemente es muy baja (< 1
/1.000.000), habiendo descritos en la literatura tan solo algunos
cientos de casos. El cuadro clínico puede ser muy polimorfo,
afectando a tejidos ectodérmicos y mesodermicos, con manifestaciones
cutáneas, orales, dentales, oculares y esqueléticas. Las dos
manifestaciones más frecuentes son la llamada hipoplasia dérmica focal
y la aparición de múltiples papilomas periorificiales, en cavidad
oral, faringe o laringeLa trasmisión es dominante ligada al X,
produciéndose habitualmente la muerte intra-útero de los pacientes
varones afectos, si bien hay casos descritos en varones con
supervivencia como consecuencia de un mosaicismo. El síndrome esta
causado por una mutación heterozigotica en el gen PORCN (gen homólogo
de la porcupina de la Drosophila) localizado en el cr Xp11.23, que es
regulador de la vía de señalización Wnt. El 90% de los pacientes
afectos son mujeres y solo el 5% de estas mujeres afectas, lo son por
herencia de la mutación, siendo el 95% de los casos mutaciones de
novo.Los datos clínicos más habituales es la aparición de áreas de
hipopigmentación cutáneas, siguiendo las líneas de Blaschkoid, junto a
atrofia de la piel, con aparición de teleangiectasias y de pápulas
amarillentas secundarias a la herniación del TCS en la dermis. Las
lesiones papilomatosas a menudo son múltiples en la cavidad oral,
faringe o laringe, no parecen estar relacionadas con el HPV y pueden
causar complicaciones obstructivas.Se presenta los datos
morfológicos detectados en la biopsia cutánea de esta afección así
como la morfología de lesiones papilomatosas en amígdala lingual y
palatina en una niña de 9 años de edad, afecta por este síndrome y sin
antecedentes familiares del síndrome. A nivel cutáneo destaca
la practica ausencia de tejido dérmico con irrupción de la grasa
hipodérmica prácticamente hasta la altura de la epidermis. A nivel de
la dermis papilar aparece un incremento en el número de capilares
dérmicos que recuerdan la imagen del angioma serpiginoso. Por otra
parte los papilomas múltiples en las amígdalas linguales y palatinas
muestran una proliferación epitelial escamosa de tipo papilomatoso
bajo la que subyace un tejido linfoide sin anomalías estructurales. La
detección del genoma viral HPV (28 cepas) mediante la metodología
Anyplex arrojo resultados negativos en los papilomas múltiples
presentes en esta enferma pediátrica
Identificadores:
http://rodrigo.uv.es/uv_da_001_0009
|
|
|
|
Veure al catàleg Trobes
|
Aquest element apareix en la col·lecció o col·leccions següent(s)
Mostra el registre complet de l'element


Per veure més documents utilitze el menú de l'esquerra
